
Featured Speakers

Raman Sumathy
ExxonMobil Research and Engineering Center, Clinton, New Jersey, USA

Denis Jacquemin
Chimie et Interdisciplinarité: Synthèse, Analyse, Modélisation · Universite de Nantes, France
The QUEST Database of Highly-Accurate Excitation Energies and Properties
The QUEST Database of Highly-Accurate Excitation Energies and Properties
Denis Jacquemina,b
aNantes Université, CNRS, CEISAM UMR 6230, F-44000, Nantes, France
bInstitut Universitaire de France, 75005, Paris Cedex 5, France
During this talk, I will describe efforts made during the last few years to create a large database of ultra-accurate reference values for electronic excited states: the QUEST database.[1] The database encompasses vertical transition energies,[1,2] oscillator strengths,[3] dipole moments,[3] excited-to-excited transition probabilities,[4] two-photon absorption strengths,[5] and geometries[6] for a large set of small and medium-sized molecules, as well as organic chromophores.[1,2,7] Excited states of various nature (p-p*, n-p*, double excitation, Rydberg, singlet, doublet, triplet, quartet…) have been considered. Most reference values have been obtained using an incremental strategy which consists in combining high-order coupled cluster and selected configuration interaction calculations using increasingly large diffuse basis sets in order to reach high accuracy. This allowed producing theoretical best estimates (TBEs) with the aug-cc-pVTZ basis set for each of these transitions, as well as basis set corrected TBEs (i.e., near the complete basis set limit) for some of them. It should be noted that the QUEST database does not rely on any experimental values, avoiding potential biases inherently linked to experiments and facilitating theoretical cross comparisons. In a second part of the talk, I will show how the TBEs/aug-cc-pVTZ have been employed to benchmark a large number of (lower-order) wave function methods such as CIS(D), ADC(2), CC2, STEOM-CCSD, CCSD, CCSDR(3), CCSD(T)(a)*, CCSDT-3, ADC(3), CC3, CASPT2, NEVPT2, and so on (including spin-scaled variants). The performances of CC4[8] and the specific case of double excitations[9,10] will be discussed as well.
[1] P. F. Loos, M. Boggio-Pasqua, A. Blondel, F. Lipparini and D. Jacquemin, J. Chem. Theory Comput. 21 (2025) 8010. Database freely available on https://github.com/pfloos/QUESTDB.
[2] M. Véril et al. WIREs Comp. Mol. Sc. 11 (2021) e1517.
[3] A. Chrayteh, A. Blondel, P. F. Loos and D. Jacquemin J. Chem. Theory Comput. 17 (2021) 416.
[4] J. Sirucek, B. Le Guennic, Y. Damour, P. F. Loos and D. Jacquemin J. Chem. Theory Comput. 21 (2025) 4688.
[5] C. Naim, R. Zalesny and D. Jacquemin J. Chem. Theory Comput. 20 (2024) 9093.
[6] S. Budzak, G. Sclamani and D. Jacquemin J. Chem. Theory Comput. 13 (2017) 6237.
[7] I. Knysh, F. Lipparini, A. Blondel, I. Duchemin, X. Blase, P. F. Loos and D. Jacquemin J. Chem. Theory Comput. 20 (2024) 8152.
[8] P. F. Loos, D. A. Matthews, F. Lipparini and D. Jacquemin J. Chem. Phys. 154 (2021) 221103.
[9] P. F. Loos, M. Boggio-Pasqua, A. Scemama, M. Caffarel and D. Jacquemin J. Chem. Theory Comput. 15 (2019) 1939.
[10] F. Kossoski, M. Boggio-Pasqua, P. F. Loos and D. Jacquemin J. Chem. Theory Comput. 20 (2024) 5655.

Michelle Coote
Flinders Institute for Nanoscale Science & Technology, Flinders University, Adelaide, Australia

Joseph Indekeu
Department of Physics, KU Leuven, Belgium

Stefan Knecht
Group Quantum Chemistry Research, Algorithmiq Ltd., Helsinki, Finland

Fernando Buendia
Department of Chemical and Biomolecular Engineering, Faculty of Engineering, National University of Singapore

Pham Le Nhan
Flinders Institute for Nanoscale Science & Technology, Flinders University, Adelaide, Australia

Phan Thi Ngoc Loan
Department of Physics, Ho Chi Minh City University of Education

Trinh Xuan Hoang
Institute of Theoretical Physics, Vietnam Academy of Science and Technology

Nguyen Ngoc Linh
Faculty of Materials Science and Engineering, Phenikaa University

Nguyen Minh Tri
Ho Chi Minh City University of Technology (HCMUT), Ho Chi Minh City, Vietnam

Phan Huu Nghia
Department of Health Sciences, Can Tho University

Truong Dinh Hieu
Duy Tan University
Oxidation of metazachlor herbicide by sulfate radical anion in gas and water: Mechanism, kinetics, and toxicity evaluation
Dinh Hieu Truong1,2,3
1Institute of Research and Development, Duy Tan University, Da Nang 550000, Viet Nam
2School of Engineering and Technology, Duy Tan University, Da Nang 550000, Viet Nam
3University of Sciences, Hue University, Hue City 530000, Viet Nam
ABSTRACT
In recent years, pesticide use has increased significantly due to their effectiveness in protecting crops from pests. This leads to several serious environmental problems that negatively impact ecosystems and human health. Metazchlor (MTZ) is a widely used herbicide for controlling annual grasses and broad-leaved weeds. Therefore, MTZ pesticide residues have been detected in many regions worldwide. This study investigated oxidation of MTZ initiated by sulfate radical anion (SO4●-) using density functional theory at the M06-2X/6-311++G(3df,3pd)//M06-2X/6-31+G(d,p) level of theory. Three mechanisms were considered: hydrogen abstraction (Abs), radical addition (Add), and single electron transfer (SET). All oxidation reactions were examined in the gas and water phases at temperatures ranging from 253 to 323 K and 283 to 323 K, respectively. The initial products would be reacted with common oxidizing agents and then tested for ecotoxicity using the ECOSAR (Ecological Structure–Activity Relationships) model[1] for aquatic organisms. As a result, the degradation of MTZ in both gas and water phases was favourable and spontaneous, with very high rate constants of 1.51 × 1013 and 4.50 × 1010 M-1 s-1, respectively. The gas-phase degradation was determined to be a selective process that occurred primarily via an Abs reaction at the H24 hydrogen atom belonging to the methyl group (C13). On the contrary, multiple Add and Abs pathways in water significantly contributed to oxidation, thereby making the process non-selective. These findings highlight the effectiveness of SO4●−-based advanced oxidation processes for MTZ removal in different environmental phases.
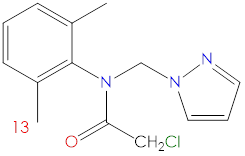
Figure 1. 2D structure of metazachlor
References
- EPA | ECOSAR. 2022 Ecological Structure Activity Relationships (ECOSAR) Predictive Model | US EPA. V2.2.

Nguyen Duc Long
Simulation in Materials Science Research Group, Science and Technology Advanced Institute, Van Lang University

Long Van Duong
Science and Technology Advanced Institute, Van Lang University, Ho Chi Minh City

Le Van Lich
Center for Materials Innovation and Technology, VinUniversity, Ha Noi

Do Thi Mai Dung
Unit of Computation and AI Application Research, Faculty of Pharmaceutical Chemistry and Technology, Hanoi University of Pharmacy

Trinh Le Huyen
Danang University of Technology, University of Danang

Nguyen Quang Trung
Danang University of Technology, University of Danang

Nguyen Minh Phuong
Center for Computational Science, Hanoi National University of Education
Nguyen Van Gia Bao
University of Science and Technology, Da Nang, Viet Nam

Bui Quang Thanh
University of Sciences, Hue University, Hue city, Vietnam